Building a complex system containing a protein (especially a metalloprotein) that contains small molecules that act as co-factors and a docked ligand can be a very challenging procedure. In order to make things easier I created this tutorial using only free and open access resources, such as Antechamber, acpype and GROMACS.
This tutorial aspires to help each user that has to perform this difficult task in the future and therefore serve the community as a free educational resource. Nonetheless, it serves an additional role. It can be referenced as a public protocol for the system preparations of the short MD simulations that followed the docking of p-cymene into each target protein to assess the stability of the protein-ligand complex, for the MSCA-funded project Waste2Drug. In this way, the reproducibility of the results and the Open Science principles of the project is ensured.
This tutorial assumes that you already have a prepared protein and ligand file. Crystal structures deposited in RCSB may have a lot of problems, such as missing hydrogens, missing side-chains, even whole missing residues and protein loops. Moreover, the specific pH of the system that you want to simulate should be taken into account for the protonation of specific residues (such as histidines). Similar manipulations (especially taking care of the protonation) should be considered also for the ligand (and any cofactors that might be present and important for your system). Finally, anything present in the opdb that it is not relevant for your simulation (such as cocrystallized water molecules in some cases or additives) should be removed from the final, prepared structure.
For this project, I used the Enalos Asclepios KNIME tools, developed by NovaMechanics Ltd for the protein and ligand preparation. An alternative that is free for academic use is CHARMM-GUI. However, there are several either paid or free options that you may use for your system’s preparation (the system preparation will not be covered by this tutorial).
Moreover, prior to the MD simulations that will be described in this tutorial, the ligand, p-cymene was docked into the binding pocket of 50 possible target proteins. The coordinates that will be used for the ligand are the output of the docking calculation. Nevertheless, this tutorial may be generalized for any protein-metal-cofactor-ligand complex that one wants to simulate.
For this tutorial, you will need an access to a machine where you can run the calculations (most probably an efficient machine with a Linux environment or access to an HPC). However, the parametrization of the ligands and the cofactors doesn’t need powerful computational resources. In my case, it was perfromed on a WSL (Windows Subsystem for Linux) on my laptop where I had set up a conda environment with Antechamber and acpype. A very useful tutorial on how to set up this environment can be found here.
Part 1: Prepare Topologies
For this work, we will use the AMBER99SB forcefield for the protein and the General Amber ForceField (GAFF) for the ligands, which is a forcefield compatible with the AMBER forcefields for proteins. In general, it is important not to mix and match forcefields and to always use compatible forcefields for different types of molecules, such as proteins, nucleic acids, small molecules etc.
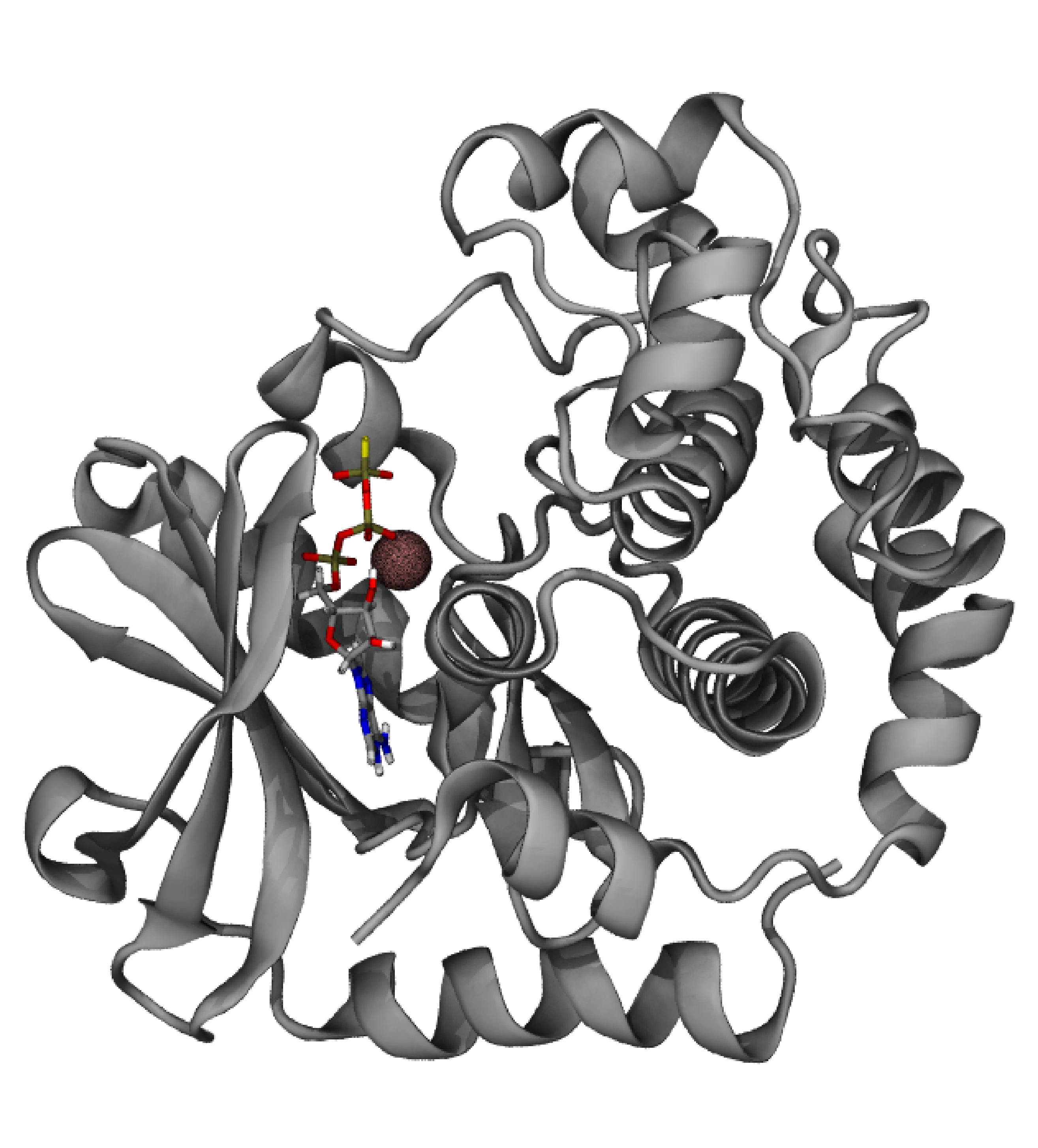
First we will need to separate the protein from the ligand and the cofactor in different structure files. The protein that we will use is the MAP2K1 (PDB ID: 3EQC), complexed with an ATP-analogue (ATP-γ-S), Mg2+ and an inhibitor. The AMBER99SB forcefield contains the parameters for all standard aminoacids and the Mg2+ ion. We will also need to keep the ATP analogue that is the physiological ligand that activates MAP2K1 but we will discard the inhibitor, as we will use our ligand, p-cymene, that was docked in the inhibitor’s pocket. In order to proceed, we need to have in different files the prepared apo proten structure (apo.pdb) that contains only standard residues and Mg2+, the ATP-analogue in .mol2 form (cofactor.mol2) and our ligand - here, the output of our docking study - again in .mol2 form (p-cymene.mol2).
-
We will first prepare the cofactor topology. In order to do so we will use the acpype code that calls Antechamber and yields small organic molecules GAFF topologies in gromacs format. In our linux environment we will do:
acpype -i cofactor.mol2After a short while, a folder will have been created in your working directory, that will contain all the relevant files that you need for the cofactor’s topology. The files that we will need are:
- cofactor_GMX.gro: the gromacs structure file for the cofactor (in this case, the ATP analogue)
- cofactor_GMX.itp: the gromacs topology file for the cofactor and
- posre_cofactor.itp: the position restraints for the cofactor.
We will follow the same procedure for the ligand:
acpype -i p-cymene.mol2and we will get the equivalent structure, topologies and restraints files.
-
Then we will prepare the protein/Mg2+ topology using the standard gromacs procedure:
gmx pdb2gmx -f apo.pdb -o apo_processed.gro -ter -ignhAfter the execution of the command we will be asked for the forcefield that we want to use (in this case the AMBER99SB forcefield), the water model (in this case TIP3P) and the type of termini that should be used in order to cap the system. In our case, the protein was already capped through protein preparation and GROMACS recognised the cappings, so no question about termini was asked. If you omit -ignh that ignores hydrogens and places new ones, it is almost certain that you will get an error, as GROMACS will not recognise the naming convention of your hydrogens.
Part 2: Combine structures and topologies
-
The next step is to combine the structures in a single .gro file for your complex. Copy the apo_processed.gro into a new file called complex.gro:
cp apo_processed.gro complex.groOpen the complex.gro with a text editor and copy the coordinates of cofactor.gro in the end:
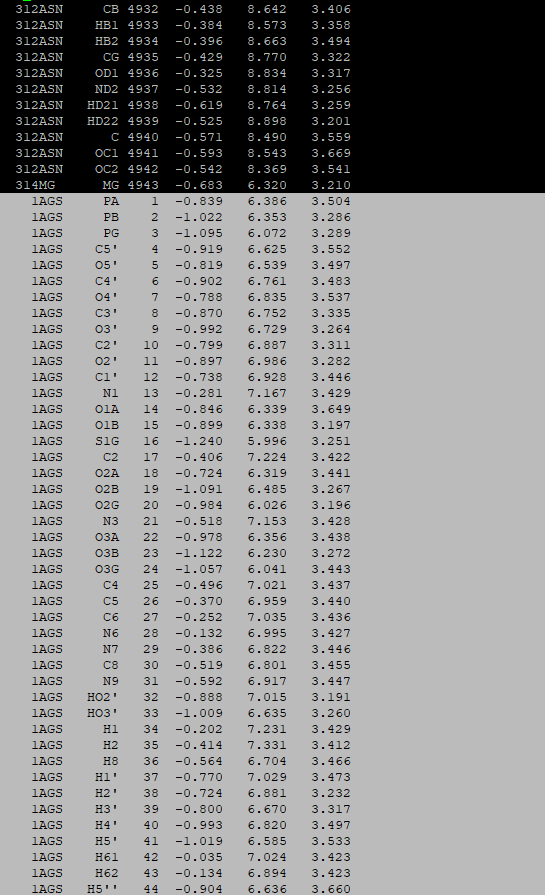
Do the same with the p-cymene.gro:
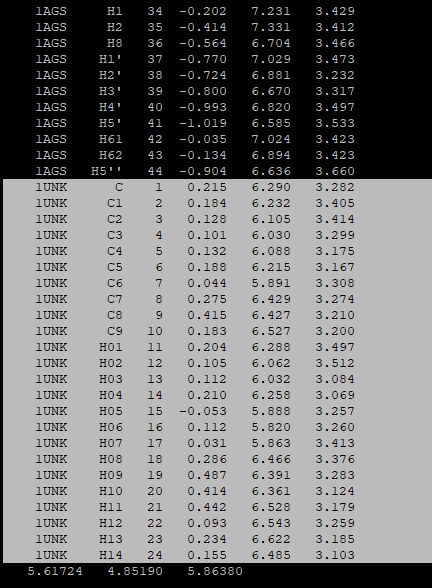
After having copied the coordinates of both the cofactor and the ligand, you will need to adjust the number of coordinates in the beginning of the complex.gro file. In our case it was:
4943 (the number in apo_processed.gro) + 44 (the number in cofactor.gro) + 24 (the number in the p-cymene.gro) = 5011 in the final complex.gro file -
Now we will have to build the topology of the system, topol.top, by combining the cofactor and ligand topologies and position restraints with those of our system. We will use
#includestatements inside the topol.top file that was already created by thepdb2gmxcommand, in order to include the .itp files for our ligands, created by the acpype and antechamber. However, some manipulation needs to be made when including several different topologies inside the topol.top file, because the#includestatements must be made with the correct order, and no [ atomtypes ] should be placed after any [ moleculetype ] directive. If we have a look inside the cofactor_GMX.itp, we will see that it starts with an [ atomtypes ] and is followed with a [ moleculetype ] directive.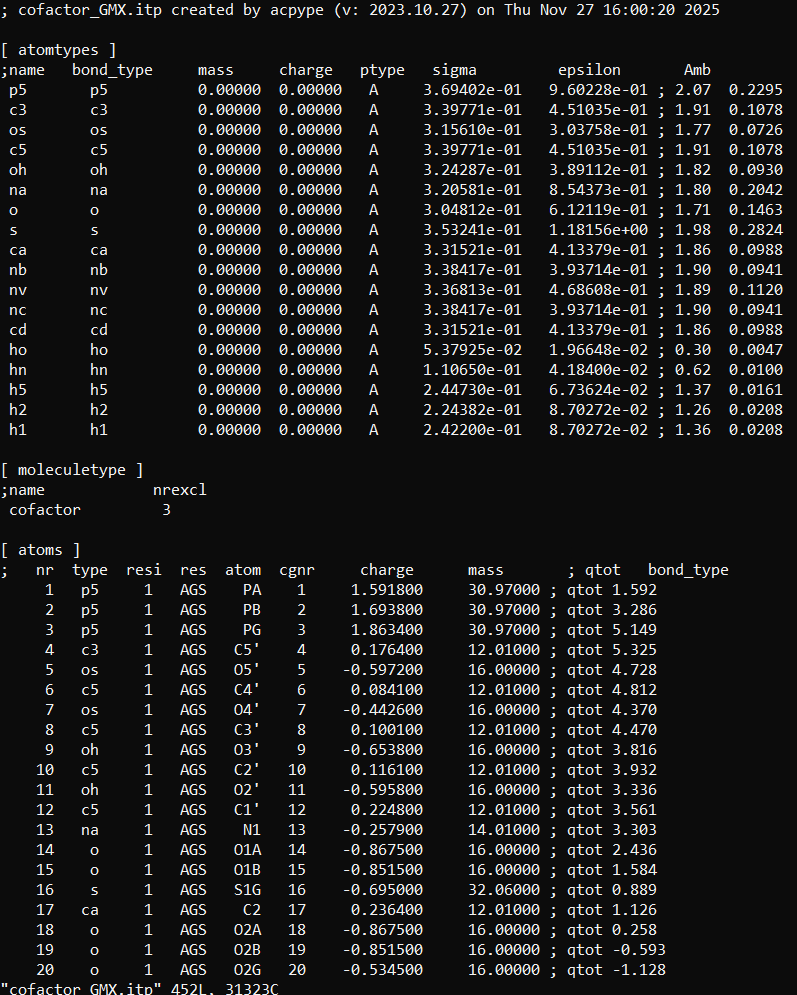
Exactly the same happens inside the p-cymene_GMX.itp file. Thus, if we consecutively include the cofactor and ligand topologies inside the topol.top file like that:
; Include ligands topology #include "cofactor_GMX.itp" #include "p-cymene_GMX.itp"we will inevitably have an [ atomtypes ] directive (that of p-cymene_GMX.itp) be placed after the [ moleculetype ] of the cofactor_GMX.itp and this will result into a GROMACS error.
In order to avoid this problem we will do a trick. We will copy the cofactor_GMX.itp to a file named cofactor.itp and we will keep only what is before the [ moleculetype ] in the original .itp file, in this case it is only the [ atomtypes ] directive. Then, we will rename the cofactor_GMX.itp to cofactor_moleculetypes.itp and we will keep only whatever is from the [ moleculetype ] directive and after. Practically, we will have divided the cofactor_GMX.itp into two different files. We will do exactly the same with the p-cymene_GMX.itp.
Now, we can include only the atom types of our ligands by placing the following exactly after the general forcefield parameters inside the topol.top file:
; Include ligands topology #include "cofactor.itp" #include "p-cymene.itp"It is wise to respect the order that the different molecule types appear in your complex.gro file when building topologies, in order to avoid future errors. Here we copied first the cofactor and thεn p-cymene after the protein inside the complex.gro file and in the topology we did the same; we first included the atomtypes for the cofactor and then for p-cymene using include statements.
-
For the next step, we will have to include the rest of the cofactor’s and ligand’s topology inside our central topol.top file. To do so, we will start with the cofactor, and right after the
; Include ligands topologyblock, we will include the molecule types for the co-factor:;Include Molecule types for co-factor #include "cofactor_moleculetypes.itp"However, we will not place right after that the equivalent file for the p-cymene, because we also need to include the position restraints for the co-factor, that must be placed exactly after the [ moleculetype ] directive for this moelcule and before any other [ moleculetype ] directive. Thus, after the
;Include Molecule types for co-factorblock we will copy the following:; co-factor position restraints #ifdef POSRES_LIG1 #include "posre_cofactor.itp" #endifThis chunk of code means that if -POSRES_LIG1 appears inside a gromacs protocol file (.mdp), the cofactor will be restarined. This is useful for the equi8libration process that will follow after the system setup. After finishing with the topology and position restrains of the cofactor, we will do the same for the ligand:
;Include Molecule types for p-cymene #include "p-cymene_moleculetypes.itp" ; p-cymene pos restraints #ifdef POSRES_LIG2 #include "posre_p-cymene.itp" #endifAfter that, the protein topology should appear in the topol.top file, as created by the
pdb2gmxcommand. In our case, GROMACS treated the protein as one chain and the Mg2+ ion as another chain, so the next block in our topol.top file reads:; Include chain topologies #include "topol_Protein_chain_A.itp" #include "topol_Ion_chain_A2.itp"and then the water and ions topologies follow.
-
In the end of our topol.top file we will find the [ molecules ] directive. It will start with one moelcule of our protein chain A (the protein), then one moelcule of our ion chain 2 will follow (the Mg2+ ion) and then we will have to manually add the cofactor and the ligand:
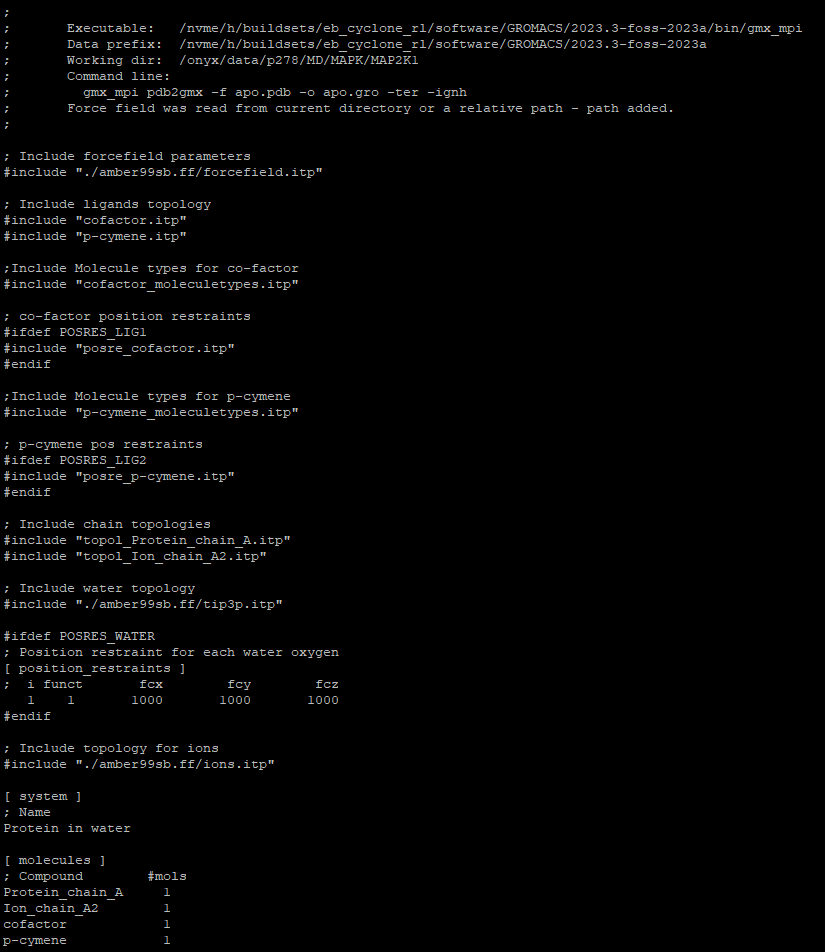
[ molecules ] ; Compound #mols Protein_chain_A 1 Ion_chain_A2 1 cofactor 1 p-cymene 1Finally, the altered topol.top file should look like this:
Now that the system is built you can follow the regular procedure for MD simulations with GROMACS. For example, you may follow this tutorial provided by the Lemkul Lab, from the Solvation part and on.
It is very possible that you will encounter an error due to “too many warnings” when you perform the “Adding Ions” step. You should carefully examinate the warnings and check if they are harmless. If they are, you might suppress the error by adding -maxwarn 1 (or 2 etc) to your gmx commands. For example, in my case the same type of atoms was defined two times, both in the topology of the cofactor and in that of p-cymene. Since after carefully checking the topologies I confirmed that they are indeed the same type of atom and overriding the second definition would not cause a problem, I chose to suppress the error by increasing the tolerance in the maximum warnings.