Working with metal-containing proteins is often very complicated, especially when no standard focefields are available for treating this specific type of metal and most importantly, the protein residues that are bonded to it. Luckily, a group of researchers form Italy published a modified AMBER forcefield that was optimized specifically for Zn proteins, first treating HIS and CYS zinc-binding residues and then extending it also for ASP and GLU.
In this work we will use the work of Marina Macchiagodena et al. in order to prepare a system for MD simulation, containing Zn and a ligand. They have also worked on cadmium-containing proteins, more details you can find here.
So, what you need to do first is to download the gromacs parameter files from the supporting info of the two papers concerning Zn mentioned above. If you decompress the .zip files, you will see a list like that below:

If you open the README file you will be directed to the corresponding gromacs page that explains how to add an external residue to a local copy of a forcefield. We will take this process step by step.
- First, you will need to create a local copy of an amber forcefield, copying it from your gromacs directory to your working folder.
⚠️ Attention: If you work on an HPC, it is better to do these manipulations with a conda version of gromacs. This is suggested because although you will be able to copy the forcefield folder from where it is loaded in the HPC system to your working directory, the residuetypes.dat will be read from where all the forcefields reside, and most probably, you won't be able to change it in your HPC system. However, if you create a conda environment with gromacs installed, you may directly change this file. This step is crucial for adding new bonded residue types.
The forcefields should be located somewhere in the
share(orliborbin) folder of the conda environment that you used. In order to easily find it do:echo "$CONDA_PREFIX"and the location of your conda environment will be printed. Go there with
cdand then search for thegromacs/topdirectory, most probably inside thesharefolder of your environment. - Then, you simply need to copy the contents of each file listed in the picture above to the relevant file in your local copy of the amber forcefield. For example, you will copy this from the
ffnonbonded.itpof the .zip file you downloaded and copy it to the end of theffnonbonded.itpof your local amber forcefield. My suggestion is to delete the Zn entry, since it already exists in your amber forcefield and it might trigger a warning.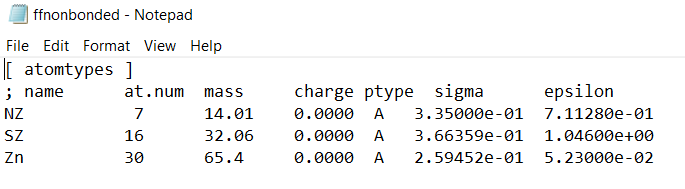
- In the
aminoacids.rtpignore the first part illustrated in the picture below, since it already exists in your local copy and multiple entries might cause errors.
-
Do the same for the rest of the files (
ffnonbonded.itp, ffbonded.itp, atomtypes.atp, aminoacids.rtp, aminoacids.hdb). For theresiduetypes.datyou will have to change the file in your gromacstopdirectory (it doesn’t work with a local copy). -
After finishing the first AMBER forcefield upgrade, containing parameters for HIS and CYS, do the same procedure with the second one, containing parameters for ASP and GLU.
-
Now it’s time to process your pdb file in order to make it work with
pdb2gmx. First you must investigate your file with a pdb viewer (I am using the open source version of Pymol ) and check which residues are bound to Zn. Take notice of the numbers of the relevant CYS, HIS, ASP and GLU. -
Next, open your pdb with a text editor and change those residues accordingly: rename CYS to CYZ, HIS to HDZ or HEZ, depending on its protonation (HDZ for HID and HEZ for HIE), GLU to GLZ and ASP to ASZ. Select the oxygen interacting with the zinc ion and rename it to OZ.
-
After changing the interacting residues accordingly, locate the Zn ions in your pdb file. Make sure that their residue is plain ZN and not ZNXX (in my case it was named ZN10 and I got an error for uknown residue).
- Another issue I encountered, is that if your ZN residues are randomly placed inside your pdb, gromacs fails during the
pdb2gmxprocess with the following error:Fatal error: The residues in the chain GLU1--VAL439 do not have a consistent type. The first residue has type 'Protein', while residue ZN1 is of type 'Ion'. Either there is a mistake in your chain, or it includes nonstandard residue names that have not yet been added to the residuetypes.dat file in the GROMACS library directory. If there are other molecules such as ligands, they should not have the same chain ID as the adjacent protein chain since it's a separate molecule. - A way to bypass this problem is by reordering your pdb file. In my case. I also had a ligand (resname UNK), so I decided to put all the ZN ions in the end of the protein just before the ligand. To do so, execute the following commands.
grep 'Zn' system.pdb > Zn.pdb && grep -v 'Zn' system.pdb > system_no_Zn.tmp && mv system_no_Zn.tmp system.pdbin order to remove the ZN ions from your system file and write two seperate files; one containing everything except Zn (named again
system.pdb) and one with only the Zn ions (namedZn.pdb) - Then we will write a new .pdb file by placing the Zn ions exactly before our ligand.
awk '/PATTERN/ && !done { system("cat Zn.pdb"); done=1 } { print }' system.pdb > systemZn.pdbreplace PATTERN with what is appropriate for your case. In my case it was “HETATM 7013 C UNK”, i.e., the first atom of my ligand. You may also do this by hand, by opening the system.pdb file and pasting the ZN residues in an appropriate place (preferably, right after the terminal residue of your protein).
- The last step is to reorder and renumber the pdb file, because after all these text modifications the numbering will not be correct. The openbabel package is needed in order to reorder and renumber the file.
obabel -ipdb systemZn.pdb -opdb -O final.pdb -
Now open your final.pdb and make sure that the ZN residues belong to a different chain from that of your protein. If not, change the Zn chain from e.g. A to B.
- Now you are ready to build your topology with:
gmx pdb2gmx -f final.pdb -ter -ignhWhen prompted, choose your local copy of the forcefield (normally the option 1)